BACKGROUNDPhylogenetic trees are hierarchical constructions used for representing the inter-relationships between organic entities. They are the commonest device for representing evolution and are important to a variety of fields throughout the life sciences.
The manipulation of phylogenetic trees-in phrases of including or eradicating tips-is usually carried out by researchers not simply for causes of administration but additionally for performing simulations to be able to perceive the processes of evolution. Despite this, the commonest programming language amongst biologists, R, has few class constructions nicely suited to those duties.
RESULTSWe current an R package that incorporates a brand new class, known as TreeMan, for representing the phylogenetic tree. This class has an inventory construction permitting phylogenetic trees to be manipulated extra effectively. Computational operating occasions are diminished as a result of of the prepared means to vectorise and parallelise strategies.
Development can be improved as a result of fewer strains of code being required for performing manipulation processes.CONCLUSIONSWe current three use cases-pinning lacking taxa to a supertree, simulating evolution with a tree-growth mannequin and detecting vital phylogenetic turnover-that show the brand new package’s velocity and simplicity.
Background
Phylogenetic trees are hierarchical constructions used for representing the inter-relationships between organic entities. They are the commonest device for representing evolution and are important to a variety of fields throughout the life sciences. The manipulation of phylogenetic trees—in phrases of including or eradicating ideas—is usually carried out by researchers not simply for causes of administration but additionally for performing simulations to be able to perceive the processes of evolution. Despite this, the commonest programming language amongst biologists, R, has few class constructions nicely suited to those duties.
Results
We current an R package that incorporates a brand new class, known as TreeMan, for representing the phylogenetic tree. This class has an inventory construction permitting phylogenetic trees to be manipulated extra effectively. Computational operating occasions are diminished as a result of of the prepared means to vectorise and parallelise strategies. Development can be improved as a result of fewer strains of code being required for performing manipulation processes.
Conclusions
We current three use instances—pinning lacking taxa to a supertree, simulating evolution with a tree-growth mannequin and detecting vital phylogenetic turnover—that show the brand new package’s velocity and simplicity.
Electronic supplementary materials
The on-line model of this text (doi:10.1186/s13104-016-2340-8) incorporates supplementary materials, which is offered to approved customers.
Background
Phylogenetic trees have been a mainstay of the R statistical software program surroundings for the reason that launch of Emmanuel Paradis’ APE package in 2002 This package launched the phylo object, an S3 class for the presentation and manipulation of phylogenetic tree knowledge within the R surroundings.
In its most simple implementation, the phylo object incorporates an inventory of three parts: an edge matrix, a vector of tip labels and an integer of the quantity of inside nodes. The use of an edge matrix facilitates phylogenetically structured statistical analyses as a result of of its comfort for producing distance, cophenetic or covariance matrices.
For this purpose the APE package’s phylo is the dominant class for phylogenetic tree illustration in R and is utilized by many well-known phylogenetic R packages (e.g. phangorn Since phylo’s first incarnation the quantity of accessible capabilities within the APE package has risen from 28 to 171 (variations 0.1–3.4), and thus far there are 147 reverse dependencies, i.e. packages on CRAN that rely upon the phylo class. More not too long ago, the phylo class has been up to date to S4 as half of the phylobase package
An edge matrix, nonetheless, results in a dependence on index referencing, resulting in sure computational eventualities during which the phylo object performs poorly: specifically, analyses that require the manipulation of the tree itself (i.e. tip and node addition/deletion). Such analyses embody simulating, evaluating, pruning, and merging trees, and calculating phylogenetic statistics equivalent to measures of phylogenetic richness and evolutionary distinctness
These have turn out to be the protect of software program options exterior to R, e.g. hindering their integration with the numerous packages in biomolecular, evolutionary and ecological research already accessible for R.
Although there are alternate options to the phylo class for phylogenetics or extra usually ‘networks’ accessible in R, these packages and lessons are not often used for phylogenetics and could lack the intuitive useful framework for manipulating evolutionary trees.
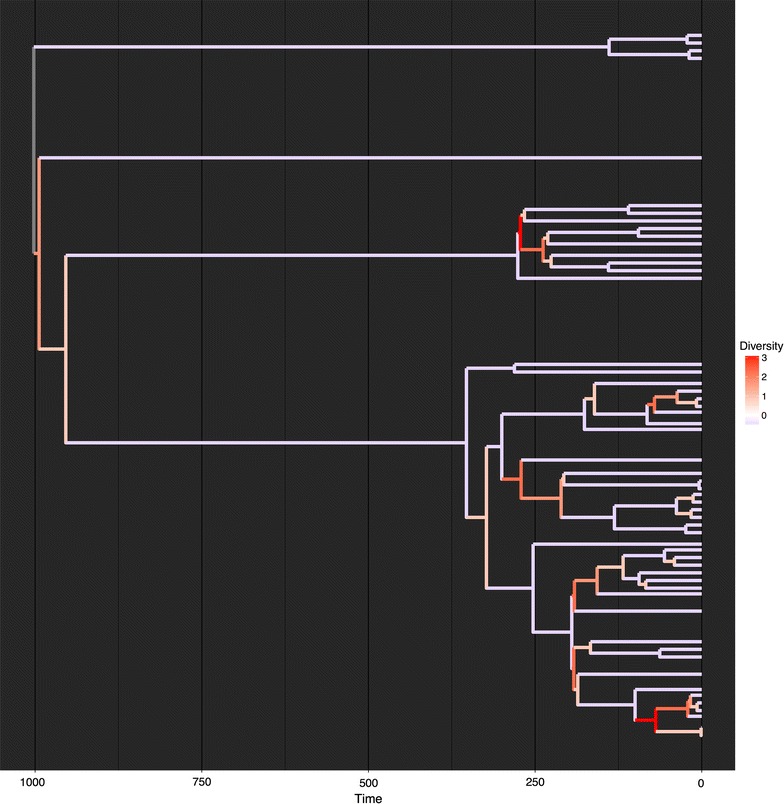
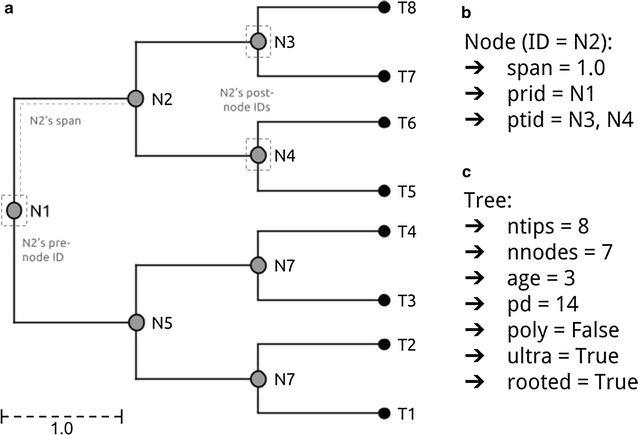
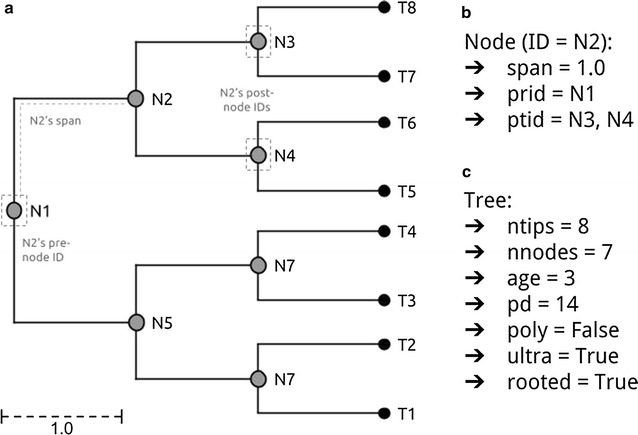
Here we current the brand new phylogenetic tree manipulation class ‘TreeMan’ (see Fig. for an overview); that is offered because the R package ‘treeman’ (N.B. the package title is all lowercase). This class is constructed round an inventory of named nodes slightly than an index-based edge matrix as is the case for the phylo class.
Using an edge matrix, every time a node is added or eliminated the brand new positions of all nodes within the matrix should be decided and the tree should be re-computed. With a node record, nonetheless, order doesn’t matter; nodes might be added and eliminated with out altering your complete tree construction.
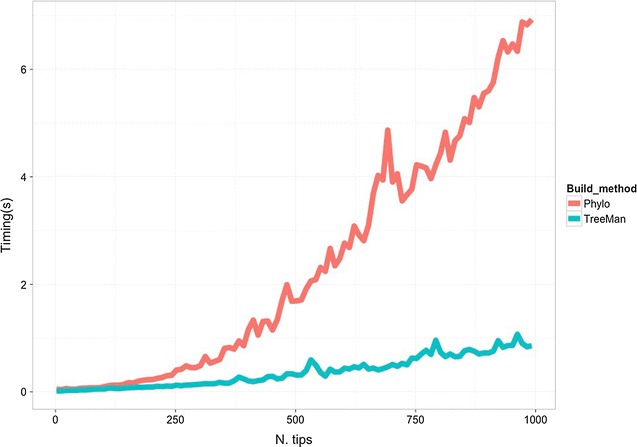
Manipulations are additionally much less depending on tree dimension as a result of all that’s required is to replace the native nodes: those who straight descend or ascend from the brand new node, changing that scale of computation time from O(N2) to O(N) (see Fig. for a comparability of rising a tree with the phylo and TreeMan lessons). Furthermore, with a node record the nodes within the tree can have distinctive IDs, which persist after insertions or deletions, permitting parts in a tree (equivalent to node labels) to be extra simply tracked throughout evaluation.
The subsequent sections of the paper describe the general construction of the brand new class, describe treeman’s naming conference, and present examples of tree manipulations that use the brand new package. The goals of treeman are to be conceptually intuitive for tree manipulation and as computationally efficient as attainable inside the R surroundings.